|
|
2.3
|
Synthese der 3-pyridyl-substituierten Pyridinium-N-phenolat-Betainfarbstoffe |
|
2.3.1
|
Synthese von 4-Amino-2,6-bis(3-pyridyl)phenol 37 |
|
2.3.1.1
|
Herstellung von 4-(3-Pyridylthioacetyl)morpholin 31[22] |
|
In einen 1l-Rundkolben mit Rückflußkühler werden 227.00 g (1.88 mol) 4-Acetylpyridin, 60.50 g (1.88 mol) Schwefel und 164.00 g (1.88 mol) Morpholin gegeben. Die Mischung wird dann lang- sam erwärmt und 12 h unter Rückfluß erhitzt. Anschließend wird die noch warme Lösung auf ca. 500 ml eines gut gerührten Eis/Wasser- Gemisches gegeben, wobei ein gelber Feststoff ausfällt. Dieser wird abfiltriert und über Di-phosphorpentoxid getrocknet. Man erhält 317.70 g (1.43 mol, 76 %) 31 als hellgelben Feststoff mit einem Schmelzpunkt von 78 °C (Lit[22]: 78 – 80 °C), der ohne weitere Reinigung weiterverwendet wird. |
|
2.3.1.2
|
Herstellung von (3-Pyridyl)essigsäure-hydrochlorid 32[22] |
|
In einem 500 ml-Rundkolben mit Rückflußkühler werden 44.40 g (0.20 mol) 4-(3-Pyridylthioacetyl)morpholin 31 und 12.00 g (0.21 mol) Kaliumhydroxid in 200 ml 95 proz. Ethanol gegeben und 72 h unter Rückfluß erhitzt. Die Mischung wird dann in 400 ml Wasser gegossen und anschließend im Rotationsverdampfer auf ca. 200 ml eingeengt (Abzug!). Es werden nochmals 200 ml Wasser zugegeben und diesmal wird die Lösung im Rotationsverdampfer fast bis zur Trockne eingeengt. Nun gibt man 200 ml 6N HCl hinzu und engt die Lösung bis zur Trockne ein. Der feste Rückstand wird dann dreimal mit je 200 ml Ethanol digeriert, und die vereinigten organischen Phasen werden im Rotationsverdampfer bis zur Trockne eingeengt. Man erhält 26.70 g (0.15 mol; 77 %) 32 in Form hellgelber Kristralle, das ohne weitere Reinigung weiter umgesetzt wird. |
|
2.3.1.3
|
Herstellung von (3-Pyridyl)essigsäureethylester 33[22], [23] |
|
Zu einer in einem 250 ml-Rundkolben mit Rückflußkühler befindlichen Mischung von 30 ml konz. H2SO4 und 70 ml Ethanol werden 26.70 g (0.15 mol) (3-Pyridyl)essigsäure-hydrochlorid 32 gegeben und das Reaktionsgemisch wird 4 h unter Rückfluß erhitzt. Die abgekühlte Lösung schüttet man auf 300 g Eis und gibt konz. Ammoniak zu, bis die Lösung alkalisch reagiert. Die wäßrige Phase wird nun vier mal mit je 300 ml Diethylether extrahiert. Die vereinigten organischen Phasen werden mit Magnesiumsulfat getrocknet und das Lösungsmittel wird im Rotationsverdampfer abdestilliert. Die zurückbleibende gelbe Flüssigkeit wird dann im Vakuum fraktionierend destilliert (120 – 122 °C, 10 Torr; Lit[22] : 121 –122 °C, 10 Torr). Man erhält 14.87 g (0.09 mol; 60 %) (3-Pyridyl)essigsäureethylester 33 als farblose Flüssigkeit, die bei +4 °C teilweise kristallisiert. |
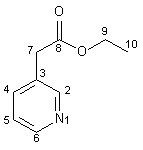
|
1 H-NMR (300 MHz, CDCl3): |
|
|
|
1.14 (t, 3H, 3J10,9 = 7.1 Hz, 10-H), 3.50 (s, 2H, 7-H), 4.05 (q, 2H, 3J9,10 = 7.1 Hz, 9-H), 7.14 (dd, 1H, 5-H) , 7.53 (m, 1H, 4-H), 8.40 (m, 2H, 2-H, 6-H). |
|
13 C-NMR (75 MHz, CDCl3): |
|
|
|
13.8 (C-10), 38.2 (C-9), 60.9 (C-7), 123.1, 129.7, 136.5, 148.3, 150.2 (C-2, C-3, C-4, C-5), 170.4 (C-8). |
|
2.3.1.4
|
Herstellung von 1,3-Bis(3-pyridyl)propan-2-on 34[24] |
|
Zu einer frisch angesetzten Natriumethanolatlösung, hergestellt aus 2.30 g (0.10 mol) Natrium und 100 ml Ethanol, werden innerhalb von 30 min 16.52 g (0.10 mol) (3-Pyridyl)-essigsäureethylester 33 getropft. Die Mischung wird 12 h bei 60 – 70 °C gerührt, dann läßt man abkühlen und gibt 80 ml konz. HCl zu. Dann erhitzt man die Lösung für 3 h unter Rückfluß. Nach dem Abkühlen gibt man 100 ml Wasser hinzu und schüttelt zweimal mit je 100 ml Chloroform aus. Die wäßrige Phase wird jetzt mit 10 proz. Natronlauge bis zur alkalischen Reaktion versetzt und anschließend dreimal mit je 100 ml Chloroform extrahiert. Die vereinigten organischen Phasen werden mit Magnesiumsulfat getrocknet und im Rotationsverdampfer bis zur Trockne eingeengt. Man erhält 2.44 g (12.00 mmol, 23 %) 34 in Form eines weißen Feststoffs mit einem Schmelzpunkt von 104 °C (Lit[24]: 103 –104 °C). |
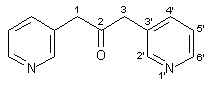
|
1 H-NMR (200 MHz, CDCl3): |
|
|
|
3.82 (s, 4H, 1/3-H), 7.25-7.32 (m, 2H, 5‘-H), 7.50-7.55 (m, 2H, 4‘-H), 8.45 (s, 2H, 2‘-H), 8.55 (d, 2H, 3J6‘, 5‘ = 3.8 Hz, 6‘-H). |
|
13 C-NMR (50 MHz, CDCl3): |
|
|
|
46.2 (C-1/3), 123.5, 129.1, 137.0, 148.7, 150.4 (C-2‘, C-3‘, C-4‘, C-5‘, C-6‘), 203.1 (C-2). |
|
2.3.1.5
|
Herstellung von 4-Nitro-2,6-bis(3-pyridyl)phenol 36 |
|
Zu einer Lösung von 4.40 g (20.80 mmol) 1,3-Bis(3-pyridyl)propan-2-on 34 und 3.40 g (20.80 mmol) Natrium-nitromalondialdehyd-monohydrat 35 in 30 ml Ethanol gibt man eine Lösung von 1.60 g Natriumhydroxid in 40 ml Wasser. Man läßt 3 d bei Raumtemperatur rühren, wobei sich das Reaktionsgemisch rot färbt. Das Ethanol wird im Rotationsverdampfer abdestilliert und der Rückstand mit 30 proz. Essigsäure neutralisiert. Es fällt ein gelber Niederschlag aus. Dieser wird abfiltriert, mehrmals mit Wasser gewaschen und schließlich mit 70 ml Ethanol heißextrahiert. Man erhält 5.18 g (17.68 mmol, 85 %) 36 in Form gelber Nadeln mit einem Schmelzpunkt von 238-239 °C. |
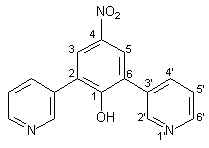
|
1 H-NMR (300 MHz, [d6]DMSO): |
|
|
|
7.51-7.57 (dd, 2H, 5‘-H), 8.04 (m, 2H, 4‘-H), 8.18 (s, 2H, 3/5-H), 8.61 (dd, 2H, 3J6‘,5‘ = 4.8 Hz, 4J6‘,4‘ = 1.5 Hz, 6‘-H), 8.82 (d, 2H, 4J2‘,4‘ = 2.2 Hz, 2‘-H). Das Phenol-H wird nicht beobachtet. |
|
13 C-NMR (75 MHz, [d6]DMSO): |
|
|
|
122.8, 125.2, 136.4 (Aromaten- und Heteroaromaten-C). Von den neun erwarteten Signalen sind aufgrund schlechter Löslichkeit und Überlagerung nur drei Signale zu erkennen. |
|
MS (FD): |
|
|
m/z (%) = |
293 (100) [M+]. |
|
IR(KBr): |
|
|
|
3450 (m, OH), 3089 (s, C-H-aromatische Schwingungen), 1588 (s, C=C-aromatische Schwingungen), 1327 (s, NO2), 1246 (s), 1111 (s, Phenol), 817 (s) und 750 (s, Deformationsschwingung in Aromaten). |
|
Elementaranalyse: |
||||
|
C16H11N3O3 (293.3) |
||||
|
Ber. (%) : Gef. (%) : |
C 65.53 C 65.24 |
H 3.78 H 3.70 |
N 14.33 N 14.35 |
|
|
2.3.1.6
|
Herstellung von 4-Amino-2,6-bis(3-pyridyl)phenol 37 |
|
1.47 g (5.00 mmol) 4-Nitro-2,6-bis(3-pyridyl)phenol 36 werden in 75 ml Ethanol und 75 ml Chloroform suspendiert und ca. 200 mg Palladium/Aktivkohle-Katalysator werden zugesetzt. Die Reaktionslösung wird dann in einer Hydrierapparatur 2 d bei Raumtemperatur und Normaldruck mit H2 umgesetzt (Wasserstoffaufnahme ca. 380 ml). Die entstandene, bernsteinfarbene Lösung wurde unter Stickstoff über eine Umkehrfritte abfiltriert und das Lösungsmittel bei Raumtemperatur im Ölpumpenvakuum abdestilliert. Nach Trocknen im Ölpumpenvakuum erhält man 1.34 g (100 %) 37 in Form eines hellbraunen Feststoffs, welcher sofort weiter eingesetzt wird. Da sich die Verbindung an der Luft zersetzt, wurde keine Analytik durchgeführt. |
|
2.3.2
|
Synthese von 4-(2,4,6-Triphenyl-1-pyridinio)-2,6-bis(3-pyridyl)-phenolat 12 |
|
2.3.2.1
|
Herstellung von 2,4,6-Triphenyl-1-[4-hydroxy-3,5-bis-(3-pyridyl)phenyl]- pyridinium-tetrafluorborat 39 |
|
1.34 g (5.00 mmol) frisch hergestelltes 4-Amino-2,6-bis(3-pyridyl)phenol 37 werden in 40 ml trockenem Methanol gelöst. Dazu gibt man 1.65 g (20.00 mmol) wasserfreies Natriumacetat und 1.98 g (5.00 mmol) 2,4,6-Triphenylpyrylium-tetrafluorborat 38. Diese Mischung wird im Wasserbad 3 h unter Rückfluß erhitzt. Die heiße, dunkelrote Lösung wird mit 30 ml Wasser versetzt, worauf ein hellgrüner Feststoff ausfällt. Man läßt über Nacht bei Raumtemperatur rühren, der ausgefallene Niederschlag wird abfiltriert, mit wenig Wasser und viel Diethylether gewaschen und im Ölpumpenvakuum getrocknet. Man erhält 2.89 g (4.51 mmol; 90 %) 39 in Form hellgrüner Kristalle mit einem Schmelzpunkt von 237-239 °C. |
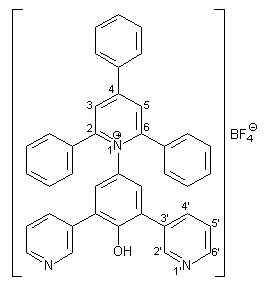
|
1 H-NMR (300 MHz, [d6]DMSO): |
|
|
|
7.42 – 7.68 (m, 19H, 3-H und 5-H des Phenolrings, Aromaten-H der 2,4,6-Phenylreste, 5‘-H), 8.27 (d, 2H 4J2‘,4‘ = 2.1 Hz, 2‘-H), 8.37 (m, 2H, 4‘-H), 8.52 (dd, 2H, 3J6‘,5‘ = 4.8 Hz, 4J6‘,4‘ = 1.6 Hz,6‘-H), 8.73 (s, 2H, 3-H und 5-H des Pyridiniumrings). Das Phenol-H-Signal wird nicht beobachtet. |
|
13 C-NMR (75 MHz, [d6]DMSO): |
|
|
|
123.5, 125.1, 127.5, 128.4, 129.0, 129.9, 130.0, 130.2, 130.7, 132.7, 133.5, 136.6, 148.7, 149.4, 156.8, 177.9 (Aromaten- und Heteroaromaten-C).Von den 20 erwarteten Signalen sind aufgrund schlechter Löslichkeit und Überlagerung nur 16 Signale zu erkennen. |
|
MS (APCI, positiv): |
|
|
m/z (%) = |
554 [M+]. |
|
IR(KBr): |
|
|
|
3430 (w, OH), 3060 (s, C-H-aromatische Schwingungen), 1622 (s, H2O), 1597 (s) und 1460 (m, C=C-aromatische Schwingungen), 1444 (s), 1401 (s), 1251 (s), 1090 (s, BF4-), 886 (s), 758 (s) und 701 (s, Deformationsschwingung in Aromaten). |
|
UV/Vis (CH3CN): |
|
|
l
max (log |
307 nm (4.581), 226 nm (4.629). |
|
Elementaranalyse: |
||||
|
C39H28BF4N3O x H2O (641.5 + 18.0 = 659.5) |
||||
|
Ber. (%) : Gef. (%) : |
C 71.03 C 71.23 |
H 4.58 H 4.79 |
N 6.37 N 6.40 |
|
|
2.3.2.2
|
Herstellung von 4-(2,4,6-Triphenyl-1-pyridinio)-2,6-bis(3-pyridyl)phenolat 12 |
|
0.64 g (1.00 mmol) Pyridiniumsalz 39 und 0.22 g (4.00 mmol) Natriummethanolat werden in 30 ml Methanol 10 min. unter Rückfluß erhitzt. Dann wird die heiße Lösung in 100 ml 10 proz. Natriumhydroxidlösung gegossen. Die dabei entstehende violette Lösung von 12 wird dreimal mit je 100 ml Chloroform extrahiert. Die vereinigten organischen Phasen werden dann mit ca. 100 ml Wasser gewaschen. Die vereinigten organischen Phasen werden dann am Rotationsverdampfer bis zur Trockne eingeengt. Der erhaltene Feststoff wird dann zuerst über P4O10 getrocknet und anschließend werden organische Lösungsmittelreste im Ölpumpenvakuum entfernt. Man erhält 0.39 g (0.71 mmol; 71 %) 12 als amorphes Pulver mit einem Schmelzpunkt von 197-198 °C |
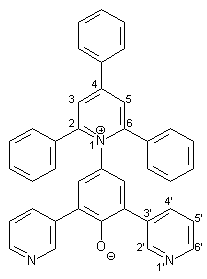
|
1 H-NMR (300 MHz, [d6]DMSO): |
|
|
|
6.93 (s, 2H, 3-H und 5-H des Phenolatrings), 7.17 (dd, 2H, 5‘-H), 7.39 – 7.65 (m, 15H, Aromaten-H der 2,4,6-Phenylreste), 7.88 (m, 2H, 4‘-H), 8.23 (dd, 2H, 3J6‘,5‘ = 4.7 Hz, 4J6‘,4‘ = 1.5 Hz, 6‘-H), 8.37 (d, 2H, 4J2‘,4‘ = 1.9 Hz, 2‘-H), 8.55 (s, 2H, 3-H und 5-H des Pyridiniumrings). |
|
13 C-NMR (75 MHz, [d6]DMSO): |
|
|
|
122.3, 124.7, 125.3, 128.3, 128.7, 128.9, 129.6, 129.8, 134.6, 135.2, 136.8, 145.7, 149.0, 153.8, 156.9, 177.9 (Aromaten- und Heteroaromaten-C). Von den 20 erwarteten Signalen sind aufgrund schlechter Löslichkeit und Überlagerung nur 16 Signale zu erkennen. |
|
MS (APCI, positiv): |
|
|
m/z = |
554 [M++1]. |
|
IR(KBr): |
|
|
|
3440 (w, OH), 3053 (s, C-H-aromatische Schwingungen), 1618 (s, H2O), 1597 (s) und 1459 (m, C=C-aromatische Schwingungen), 1459 (s), 1282 (s), 1255 (s), 814 (s), 766 (s) und 696 (s, Deformationsschwingung in Aromaten). |
|
UV/Vis (CH3CN): |
|
|
l
max (log |
590 nm (3.560), 416 nm (3.891), 302 nm (4.531), 232 nm (4.524). |
|
Elementaranalyse: |
||||
|
C39H27N3O x 2.5 H2O x 0.5 CHCl3 (553.7 + 45.0 + 59.7 = 658.4) |
||||
|
Ber. (%) : Gef. (%) : |
C 72.06 C 71.92 |
H 4.98 H 4.71 |
N 6.38 N 6.18 |
|
|
2.3.3
|
Synthese von 4-[4-Phenyl-2,6-bis(3-pyridyl)-1-pyridinio]-2,6-bis- (3-pyridyl)-phenolat 13 |
|
2.3.3.1
|
Herstellung von 2,6-Bis[(1H)-pyridinio-3-yl]-4-phenylpyrylium-tris(tetrafluorborat) 40 |
|
In einen 50 ml-Einhalsrundkolben werden 7.50 g (62.00 mmol) frisch destilliertes 3-Acetylpyridin und 3.30 g (31.00 mmol) frisch destillierter Benzaldehyd gegeben. Über einen Tropftrichter mit Druckausgleich, auf den noch ein Rückflußkühler gesetzt wird, werden dann 20 ml 54 proz. etherische Tetrafluorborsäure langsam zugetropft. Dabei fällt ein gelber Feststoff aus. Die Reaktionsmischung wird mit einem Heizpilz erwärmt, bis der gesamte Diethylether in den Tropftrichter einkondensiert ist. Die verbliebene Reaktionsmischung wird dann auf ca. 150 °C erwärmt und weitere 10 min gerührt. Nach dem Abkühlen wird die braune Reaktionsmischung mehrmals mit Diethylether digeriert und der Diethylether verworfen. Die zurückbleibende gummiartige Masse wird dann mit ca. 20 ml Aceton versetzt und 2 h bei Raumtemperatur gerührt, um die organischen Verunreinigungen herauszulösen. Der zurückbleibende Feststoff wird abfiltriert und mehrmals mit Aceton gewaschen. Der Feststoff noch einmal in 15 ml heißem Aceton gerührt, wieder abfiltriert, und die gleiche Prozedur noch einmal wiederholt. Das Produkt wird im Vakuum getrocknet. Man erhält 7.29 g (12.70 mmol; 41 %) 40 in Form gelber, fluoreszierender Kristalle mit einem Schmelzpunkt von 167 °C. |
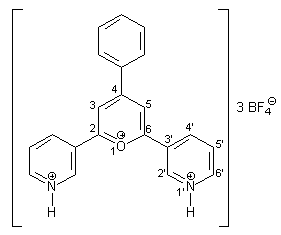
|
1 H-NMR (300 MHz, CF3COOD): |
|
|
|
7.97 (m, 2H, m-Aromaten-H des 4-Phenylrests), 8.17 (m, 1H, p-Aromaten-H des 4-Phenylrests), 8.58 (m, 2H, 5‘-H), 8.67 (m, 2H, o-Aromaten-H des 4-Phenylrests), 9.35 (m, 2H, 6‘-H), 9.43 (m, 2H, 2‘-H), 9.80 (m, 2H, 4‘-H), 10.15 (s, 2H, 3-H und 5-H). |
|
13 C-NMR (75 MHz, CF3COOD): |
|
|
|
120.5, 130.3, 132.3, 132.4, 143.7, 146.6, 148.1, 167.1 (Aromaten- und Heteroaromaten-H), 188.4 (C-6). Von den zwölf erwarteten Signalen sind aufgrund schlechter Löslichkeit und Überlagerung nur neun Signale zu erkennen. |
|
MS (FD): |
|
|
m/z (%) = |
311 (100) [M+ - BF4, -2 HBF4]. |
|
IR(KBr): |
|
|
|
3396 (w, OH), 1627 (s, H2O), 1590 (s) und 1460 (m, C=C-aromatische Schwingungen), 1419 (s), 1292 (s), 1260 (s), 1059 (s, BF4-), 961 (s), 780 (s) und 669 (s, Deformationsschwingung in Aromaten). |
|
Elementaranalyse: |
||||
|
C21H17B3F12N2O x 1.5 H2O (573.8 + 27.0 = 600.8) |
||||
|
Ber. (%) : Gef. (%) : |
C 42.25 C 41.98 |
H 3.75 H 3.36 |
N 4.33 N 4.66 |
|
|
2.3.3.2
|
Herstellung von 4-[4-Phenyl-2,6-bis(3-pyridyl)-1-[4-hydroxy-3,5-bis- (3-pyridyl)-phenyl]pyridinium-tertrafluorborat 41 |
|
Die Herstellung von 41 entspricht der Synthese von 39 (s. 2.3.2.1). Es werden 1.34 g (5.00 mmol) frisch hergestelltes 4-Amino-2,6-bis(3-pyridyl)phenol 37 in 40 ml trockenem Methanol gelöst. Dazu gibt man 1.65 g (20.00 mmol) wasserfreies Natriumacetat und 2.86 g (5.00 mmol) des Pyryliumsalzes 40. Man erhält 2.76 g (4.30 mmol; 86 %) 41 in Form farbloser Kristalle mit einem Schmelzpunkt von 192-194 °C. |
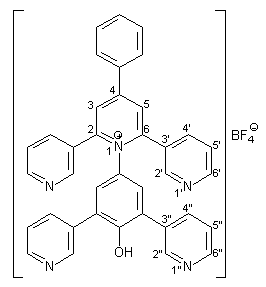
|
1 H-NMR (300 MHz, [d6]DMSO): |
|
|
|
7.43 – 7.74 (m, 11H, Aromaten-H des 4-Phenylrings, 3-H und 5-H des Phenol- rings, 5‘-H, 5‘‘-H, ), 7.95 (m, 2H, 4‘-H oder 4‘‘-H), 8.32 (d, 2H, 4J2‘,4‘ = 1.9 Hz, 2‘-H oder 2‘‘-H), 8.40 (m, 2H, 4‘-H oder 4‘‘-H), 8.55 (dd, 2H, 3J6‘,5‘ = 4.8 Hz, 4J6‘,5‘ = 1.5 Hz, 6‘-H oder 6‘‘-H), 8.66 (dd, 2H, 3J6‘,5‘ = 4.8 Hz , 4J6‘,5‘ = 1.5 Hz, 6‘-H oder 6‘‘-H), 8.78 (d, 2H, 4J2‘‘,4‘‘ = 2.0 Hz, 2‘-H oder 2‘‘-H), 8.91 (s, 2H, 3-H und 5-H des Pyridiniumrings). Das Phenol-H wird nicht beobachtet. |
|
13 C-NMR (75 MHz, [d6]DMSO): |
|
|
|
123.3, 123.6, 126.0, 128.1, 129.1, 129.6, 132.4, 136.6, 137.5, 148.9, 149.3, 149.7, 151.1, 154.3, 177.9 (Aromaten- und Heteroaromaten-C) Von den 21 erwarteten Signalen sind aufgrund schlechter Löslichkeit und Überlagerung nur 15 Signale zu erkennen. |
|
MS (APCI, positiv): |
|
|
m/z = |
556 [M+]. |
|
IR(KBr): |
|
|
|
3430 (w, OH), 3066 (s, C-H-aromatische Schwingungen), 1624 (s, H2O), 1591 (s) und 1460 (m, C=C-aromatische Schwingungen), 1401 (s), 1257 (s), 1059 (s, BF4-), 809 (s), 769 (s) und 710 (s, Deformationsschwingung in Aromaten). |
|
UV/Vis (CH3CN): |
|
|
l
max (log |
309 nm (4.558), 228 nm (4.598). |
|
Elementaranalyse: |
||||
|
C37H26BF4N5O2 x H2O (643.4 + 18.0 = 661.5) |
||||
|
Ber. (%) : Gef. (%) : |
C 67.19 C 66.88 |
H 4.27 H 4.70 |
N 10.59 N 10.31 |
|
|
2.3.3.3
|
Herstellung von 4-[4-Phenyl-2,6-bis(3-pyridyl)-1-pyridinio]- 2,6-bis-(3-pyridyl)-phenolat 13 |
|
Die Herstellung des Betainfarbstoffs 13 entspricht der Synthese von 12 (s. Kapitel 2.3.3.2). Es werden 0.64 g (1.00 mmol) Pyridiniumsalz 40 mit 0.22 g (4.00 mmol) Natriummethanolat in 30 ml Methanol umgesetzt. Man erhält 0.38 g (0.68 mmol; 68 %) Betainfarbstoff 13 in Form eines violetten Pulvers mit einem Schmelzpunkt von 198 - 200 °C. |
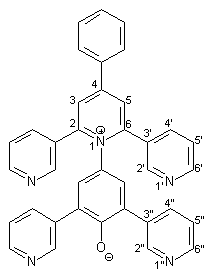
|
1 H-NMR (300 MHz, CDCl3): |
|
|
|
6.62 (s, 2H, 3-H und 5-H des Phenolatrings), 7.11 (dd, 2H, 5‘-H oder 5‘‘-H), 7.32 (dd, 2H, 5‘-H oder 5‘‘-H), 7.60 – 7.70 (m, 5H, Aromaten-H des |
|
13 C-NMR (75 MHz, CDCl3): |
|
|
|
122.4, 123.5, 126.2, 127.7, 127.9, 128.0, 129.8, 130.3, 133.1, 135.7, 136.2, 136.8, 146.7, 148.7, 148.9, 151.7 (Aromaten- und Heteroaromaten-H). Von den 21 erwarteten Signalen sind aufgrund schlechter Löslichkeit und Überlagerung nur 16 Signale zu erkennen. |
|
MS (APCI, positiv): |
|
|
m/z = |
556 [M++1]. |
|
IR(KBr): |
|
|
|
3430 (w, OH), 3037 (s, C-H-aromatische Schwingungen), 1619 (s, H2O), 1589 (s) und 1482 (m, C=C-aromatische Schwingungen), 1459 (s), 1408 (s), 1255 (s), 811 (s), 711 (s) und 709 (s, Deformationsschwingung in Aromaten). |
|
UV/Vis (CH3CN): |
|
|
l
max (log |
605 nm (3.655), 419 nm (3.891), 381 nm (3.931), 305 nm (4.603), 229 nm (4.589). |
|
Elementaranalyse: |
||||
|
C37H25N5O x 2.5 H2O (555.6 + 45.0 = 600.7) |
||||
|
Ber. (%) : Gef. (%) : |
C 73.98 C 73.87 |
H 5.03 H 5.51 |
N 11.66 N 11.24 |
|
|
2.3.4
|
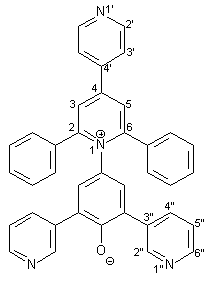
Synthese von 4-[2,6-Diphenyl-4-(4-pyridyl)-1-pyridinio]- 2,6-bis(3-pyridyl)-phenolat 14 |
|
0.54 g (2.00 mmol) frisch hergestelltes 4-Amino-2,6-bis(3-pyridyl)phenol 37 werden in 15 ml trockenem Methanol gelöst. Dazu gibt man 0.66 g (8.00 mmol) wasserfreies Natriumacetat und 0.97 g (2.00 mmol) 2,4-Diphenyl-4-[(1H)-pyridinio-4-yl]pyrylium-tetrafluorborat 42. Diese Mischung wird im Wasserbad 3 h unter Rückfluß erhitzt. 0.44 g (8.00 mmol) Natriummethanolat werden zugegeben und die Reaktionsmischung weitere 10 min. unter Rückfluß erhitzt. Dann wird die heiße Lösung in 100 ml 10 proz. Natriumhydroxidlösung gegeben. Die dabei entstehende violette Lösung von 14 wird dreimal mit je 100 ml Chloroform extrahiert. Die vereinigten organischen Phasen werden dann mit ca. 100 ml Wasser gewaschen. Die vereinigten organischen Phasen werden dann am Rotationsverdampfer bis zur Trockne eingeengt. Der erhaltene Feststoff wird zuerst über P4O10 getrocknet und anschließend werden organische Lösungsmittelreste im Ölpumpenvakuum entfernt. Man erhält 0.74 g (0.13 mmol; 67 %) 14 als amorphes, violettes Pulver mit einem Schmelzpunkt von 213 - 215 °C. |
|
1 H-NMR (300 MHz, CDCl3 ): |
|
|
|
6.50 (s, 2H, 3-H und 5-H des Phenolatrings), 7.06 (dd, 2H, 5‘‘-H), 7.30 – 7.46 (m, 10H, Aromaten-H der 2,6-Phenylreste), 7.72 (dd, 3J3‘,2‘ = 4.5 Hz, 4J3‘,3‘ = 1.6 Hz, 3‘-H ), 7.89 (d, 2H, 4J2‘‘,4‘‘ = 1.9 Hz, 2‘‘-H), 8.04 – 8.16 (m, 4H, 3-H und 5-H des Pyridiniumringes, 4‘‘-H), 8.24 (dd, 2H, 3J6‘‘,5‘‘ = 4.8 Hz, 4J6‘‘,4‘‘ = 1.7 Hz, 6‘‘-H), 8.86 (dd, 2H, 3J2‘,3‘ = 4.5 Hz, 4J2‘,2‘ = 1.6 Hz, 2‘-H). |
|
13 C-NMR (75 MHz, CDCl3): |
|
|
|
118.5, 121.2, 122.1, 126.3, 127.3, 127.8, 128.9, 129.3, 130.7, 133.3, 136.1, 136.7, 141.2, 146.3, 148.8, 151.5, 151.6, 157.4, 170.2 (Aromaten- und Heteroaromaten-H). |
|
MS (APCI, positiv): |
|
|
m/z = |
555 [M++1]. |
|
IR(KBr): |
|
|
|
3414 (w, OH), 3028 (s, C-H-aromatische Schwingungen), 1622 (s, H2O), 1593 (s) und 1484 (m, C=C-aromatische Schwingungen), 1462 (s), 1285 (s), 1257 (s), 818 (s), 766 (s) und 698 (s, Deformationsschwingung in Aromaten). |
|
UV/Vis (CH3CN): |
|
|
l
max (log |
629 nm (3.673), 415 nm (3.931), 386 nm (3.946), 251 nm (4.611). |
|
Elementaranalyse: |
||||
|
C38H26N4O x 2 H2O (554.7 + 36.0 = 590.7) |
||||
|
Ber. (%) : Gef. (%) : |
C 77.27 C 77.65 |
H 5.12 H 4.71 |
N 9.49 N 9.54 |
|
|
2.3.5
|
Synthese von 4-[2,4-Diphenyl-6-(4-methansulfonyl-phenyl)-1-pyridinio]- 2,6-bis(3-pyridyl)phenolat 15 |
|
2.3.5.1
|
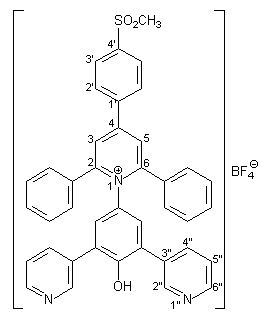
Herstellung von 2,6-Diphenyl-4-(4-methansulfonyl-phenyl)-1- [4-hydroxy-3,5-bis(3-pyridyl)-phenyl]pyridinium-tetrafluorborat 44 |
|
Analog zur Synthese von 39 (s. 2.3.2.1) wurde das Pyridiniumsalz 44 hergestellt. Es werden 1.34 g (5.00 mmol) frisch hergestelltes 4-Amino-2,6-bis(3-pyridyl)phenol 37 in 40 ml trockenem Methanol gelöst. Dazu gibt man 1.65 g (20.00 mmol) wasserfreies Natriumacetat und 2.37 g (5.00 mmol) des Pyryliumsalzes 26 (s. Kapitel 2.2.2.2). Man erhält 2.91 g (4.05 mmol; 81 %) 44 in Form hellgelber Kristalle mit einem Schmelzpunkt von 190 - 192 °C. |
|
1 H-NMR (300 MHz, [d6]DMSO): |
|
|
|
3.37 (s, 3H, SO2CH3), 7.39 – 7.63 (m, 16H, Aromaten-H der 2,6-Phenylreste, 3-H und 5-H des Phenolrings, 4‘‘-H, 5‘‘-H), 8.19 (d, 3J2‘,3‘ = 8.4 Hz, 2‘-H), 8.27 (d, 2H, 2‘‘-H), 8.53 (d, 2H, 6‘‘-H), 8.63 (d, 2H, 3J3‘,2‘ = 8.4 Hz, 3‘-H), 8.84 (s, 2H, 3-H und 5-H des Pyridiniumrings), 9.38 (s, 1H, OH). |
|
13 C-NMR (75 MHz, [d6]DMSO): |
|
|
|
123.7, 126.2, 127.7, 128.2, 128.5, 130.0, 130.4, 130.7, 132.7, 136.7, 138.4, 143.9, 148.8, 149.4, 154.0, 157.2 (Aromaten- und Heteroaromaten-H). Von den 21 erwarteten Signalen sind aufgrund schlechter Löslichkeit und Überlagerung nur 16 Signale zu erkennen. |
|
MS (APCI, positiv): |
|
|
m/z = |
632 [M+]. |
|
IR(KBr): |
|
|
|
3430 (w, OH), 3060 (s, C-H-aromatische Schwingungen), 1624 (s, H2O), 1597 (s) und 1495 (m, C=C-aromatische Schwingungen), 1453 (s), 1307 (s, SO2), 1237 (s), 1152 (s, SO2), 1057 (s, BF4-), 817 (s), 771 (s) und 705 (s, Deformationsschwingung in Aromaten). |
|
UV/Vis (CH3CN): |
|
|
l
max (log |
293 nm (4.553), 225 nm (4.616). |
|
Elementaranalyse: |
||||
|
C40H30BF4N3O3S x 2 H2O (719.6 + 36.0 = 755.6) |
||||
|
Ber. (%) : Gef. (%) : |
C 63.58 C 63.61 |
H 4.23 H 4.54 |
N 5.56 N 5.31 |
|
|
2.3.5.2
|
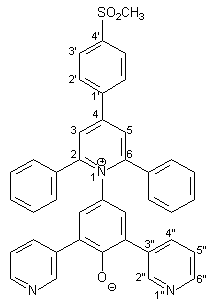
Herstellung von 4-[2,4-Diphenyl-6-(4-methansulfonyl-phenyl)-1-pyridinio]- 2,6-bis(3-pyridyl)phenolat 15 |
|
Man löst 0.72 g (1.00 mmol) des Pyridiniumsalzes 44 und 0.22 g (4.00 mmol) Natriummethanolat in 30 ml wasserfreiem Methanol und erhitzt 10 min. unter Rückfluß. Die heiße Lösung wird dann in 100 ml einer 10 proz. wäßrigen Natriumhydroxidlösung gegeben. Es bildet sich ein violetter Niederschlag, der noch 24 h bei +4 °C stehengelassen und dann abfiltriert wird. Der Niederschlag wird mit wenig Wasser und dreimal mit ja 50 ml Diethylether gewaschen. Zur Reinigung wird der Feststoff zweimal einer Heißextraktion mit je 40 ml Wasser/Methanol (2:1) unterzogen. Nach Trocknung über Diphosphorpentoxid erhält man 0.54 g (0.85 mmol; 85%) 15 in Form dunkelgrüner Kristalle mit einem Schmelzpunkt von 219 - 221 °C. |
|
1 H-NMR (300 MHz, [d6]DMSO): |
|
|
|
3.34 (s, 3H, SO2CH3), 6.91 (s, 2H, 3-H und 5-H des Phenolatrings), 7.17 (m, 2H, 5‘‘-H), 7.45 – 7.68 (m, 10H, Aromaten-H der 2,6-Phenylreste), 7.89 (d, 2H, 3J6‘,5‘= 7.6 Hz, 4‘‘-H) 8.16 (d, 2H, 3J2‘,3‘ = 8.1 Hz, 2‘-H) 8.22 (m, 2H, 6‘‘-H), 8.36 (s, 2H, 2‘‘-H), 8.57 (d, 2H, 3J3‘,2‘ = 8.1 Hz, 3‘-H) 8.66 (s, 2H, 3-H und 5-H des Pyridiniumrings). |
|
13 C-NMR (75 MHz, [d6]DMSO): |
|
|
|
122.2, 126.3, 128.0, 128.3, 129.8, 134.5, 135.3, 145.6, 149.0 (Aromaten- und Heteroaromaten-C). Von den 21 erwarteten Signalen sind aufgrund schlechter Löslichkeit und Überlagerung nur neun Signale zu erkennen. |
|
MS (APCI, positiv): |
|
|
m/z = |
632 [M++1]. |
|
IR(KBr): |
|
|
|
3400 (w, OH), 1621 (s, H2O), 1597 (s) und 1484 (m, C=C-aromatische Schwingungen), 1454 (s), 1304 (s), 1287 (s), 1149 (s, SO2), 962 (s), 772 (s) und 705 (s, Deformationsschwingung in Aromaten). |
|
UV/Vis (CH3CN): |
|
|
l
max (log |
627 nm (3.659), 415 nm (3.906), 386 nm (3.911), 286 nm (4.582), 244 nm (4.520). |
|
Elementaranalyse: |
||||
|
C40H29N3O3S x 4 H2O (631.7 + 72.1 = 703.8) |
||||
|
Ber. (%) : Gef. (%) : |
C 68.26 C 68.55 |
H 5.30 H 5.41 |
N 5.97 N 5.49 |
|
|
2.3.6
|
Synthese von 4-[2,6-Diphenyl-4-(4-nitrophenyl)-1-pyridinio]-2,6-bis- (3-pyridyl)phenolat 16 |
|
2.3.6.1
|
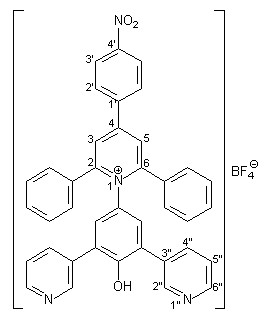
Herstellung von 2,6-Diphenyl-4-(4-nitrophenyl)-1-[4-hydroxy-3,5-bis- (3-pyridyl)phenyl]pyridinium-tetrafluorborat 45 |
|
Die Herstellung von 45 entspricht in Ansatzgröße und Durchführung der Synthese von 39 (s. Kapitel 2.3.2.1). 1.34 g (5.00 mmol) frisch hergestelltes 4-Amino-2,6-bis(3-pyridyl)phenol 37 werden in 40 ml trockenem Methanol gelöst. Dazu gibt man 1.65 g (20.00 mmol) wasserfreies Natriumacetat und 2.21 g (5.00 mmol) Pyryliumsalz 29, das nach einer Vorschrift von Gerson und Heilbronner[21] hergestellt wurde. Man erhält 3.02 g (4.40 mmol; 88 %) 45 in Form farbloser Kristalle mit einem Schmelzpunkt von 194 - 196 °C. |
|
1 H-NMR (300 MHz, [d6]DMSO): |
|
|
|
7.36 – 7.64 (m, 16H, Aromaten-H der 2,6-Phenylreste, 3-H und 5-H des Phenolrings, 4‘‘-H, 5‘‘-H), 8.25 (d, 2H, 4J2‘,4‘ = 1.6 Hz, 2‘‘-H), 8.45 (d, 2H, 3J2‘,3‘= 8.9 Hz, 2‘-H), 8.51 (dd, 2H, 6‘‘-H), 8.62 (d, 2H, 3J3‘,2‘ = 8.9 Hz, 3‘-H), 8.84 (s, 2H, 3-H und 5-H des Pyridiniumrings). Das Phenol-H wird nicht beobachtet. |
|
13 C-NMR (75 MHz, [d6]DMSO): |
|
|
|
123.6, 124.5, 128.4, 129.9, 130.5, 136.5, 149.3, 177.9 (Aromaten- und Heteroaromaten-H). Von den 20 erwarteten Signalen sind aufgrund schlechter Löslichkeit und Überlagerung nur acht Signale zu erkennen. |
|
MS (APCI, positiv): |
|
|
m/z = |
599 [M+]. |
|
IR(KBr): |
|
|
|
3429 (w, OH), 3060 (s, C-H-aromatische Schwingungen), 1626 (s, H2O), 1597 (s) und 1452 (m, C=C-aromatische Schwingungen), 1434 (s), 1349 (s, NO2-Valenz), 1249 (s), 817 (s), 762 (s) und 702 (s, Deformationsschwingung in Aromaten). |
|
UV/Vis (CH3CN): |
|
|
l
max (log |
299 nm (4.585), 227 nm (4.582). |
|
Elementaranalyse: |
||||
|
C39H27BF4N4O3 x H2O (686.5 + 18.0 = 704.5) |
||||
|
Ber. (%) : Gef. (%) : |
C 66.49 C 66.20 |
H 4.15 H 4.39 |
N 7.95 N 7.61 |
|
|
2.3.6.2
|
Herstellung von 4-[2,6-Diphenyl-4-(4-nitrophenyl)-1-pyridinio]-2,6-bis- (3-pyridyl)phenolat 16 |
|
Die Synthese des Betainfarbstoffs 16 entspricht in Ansatzgröße und Durchführung der Synthese von 15 (s. Kapitel 2.3.5.2). Es werden 0.69 g (1.00 mmol) des Pyridiniumsalzes 45 mit 0.22 g (4.00 mmol) Natriummethanolat in 30 ml wasserfreiem Methanol deprotoniert. Man erhält 0.60 g (0.87 mmol; 87 %) Betainfarbstoff 16 in Form eines violetten Pulvers, das sich ab 275 °C zersetzt. |
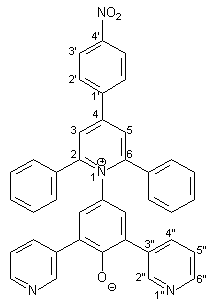
|
1 H-NMR (300 MHz, [d6]DMSO): |
|
|
|
6.91 (s, 2H, 3-H und 5-H des Phenolatrings), 7.16 (dd, 2H, 5‘‘-H), 7.37 – 7.63 (m, 10H, Aromaten-H der 2,6-Phenylreste), 7.88 (m, 2H, 4‘‘-H), 8.22 (dd, 2H, J nicht bestimmbar, 6‘‘-H), 8.35 (d, 2H, J nicht bestimmbar, 2‘‘-H), 8.42 (d, 2H, 3J2‘,3‘ = 8.8 Hz, 2‘-H), 8.58 (d, 2H, 3J3‘,2‘= 8.8 Hz, 3‘-H), 8.67 (s, 2H, 3-H und 5-H des Pyridiniumrings). |
|
13 C-NMR (75 MHz, [d6]DMSO): |
|
|
|
122.4, 128.6, 130.0, 130.5, 135.5, 149.2, 178.2 (Aromaten- und Hetero- |
|
MS (APCI, positiv): |
|
|
m/z = |
598 [M+]. |
|
IR(KBr): |
|
|
|
3402 (w, OH), 3058 (s, C-H-aromatische Schwingungen), 1618 (s, H2O), 1596 (s) und 1462 (m, C=C-aromatische Schwingungen), 1542 (s, NO2-Valenz), 1410 (s), 1344 (s, NO2-Valenz), 1289 (s), 815 (s), 769 (s) und 696 (s, Deformationsschwingung in Aromaten). |
|
UV/Vis (CH3CN): |
|
|
l
max (log |
646 nm (3.684), 414 nm (3.956), 386 nm (3.997), 295 nm (4.612), 242 nm (4.520). |
|
Elementaranalyse: |
||||
|
C39H26N4O3 x 2 H2O (598.7 + 36.0 = 634.7) |
||||
|
Ber. (%) : Gef. (%) : |
C 72.77 C 72.43 |
H 4.85 H 5.11 |
N 8.70 N 8.24 |
|
|